Transcriptomes of cells, tissues or organisms can vary widely depending on the surrounding living conditions. Our high-throughput methods for sequencing mRNA allow us to analyze whole transcriptomes.By applying different protocols in the sample processing (RNAseq, EST) and high throughput sequencing, as well as adapted bioinformatic analysis, we can answer various questions.By transcriptome data, new and rare transcripts can be identified and the annotation of the organism can be improved.The analysis of differential gene expression is also made possible by transcriptome dataWe are happy to assist you in the implementation of transcriptome projects.
RNASeq
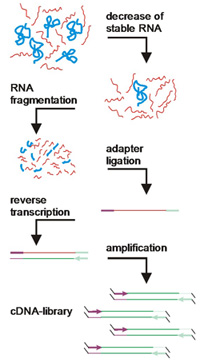
For mRNA sequencing, m the RNA is first transcribed into cDNA and can then be sequenced with all NGS technologies.However, care should be taken to reduce the very high proportion of rRNAs.The sequenced fragments are then mapped to the reference genome, with 75bp sequencing reads reflecting optimal results. The required high coverage is achieved here by a large number of individual reads. The HiSeq 1500 for large genomes, the NextSeq 500 for medium size or the MiSeq for small genomes are ideally suited for mRNA sequencing.
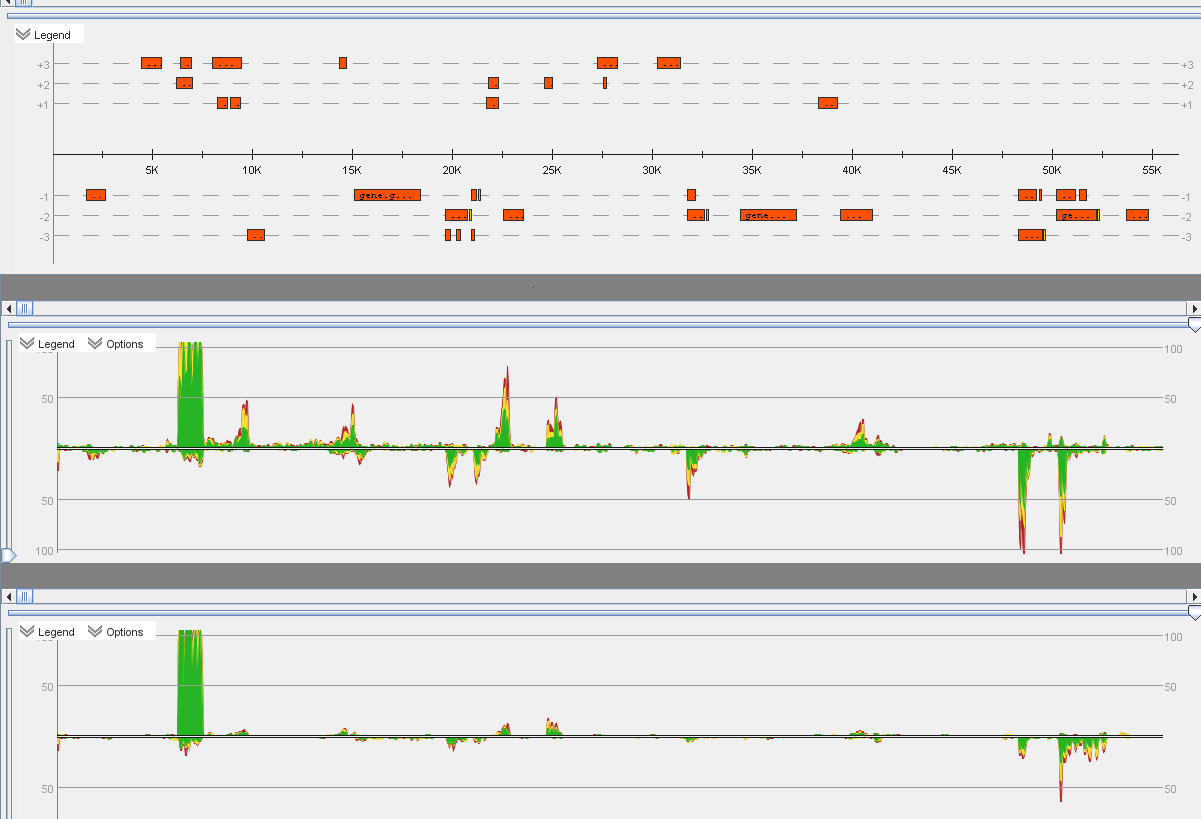
Example of an RNASeq project:
- Sequencing on the Illumina HiSeq 1500
+ Read mapping
+ Differential gene expression analysis
+ Operon detection
+ Start codon search
+ Motif search