Diverse microorganisms have properties that are of great interest for industrial or medical applications. Based on genome information, you can better understand and analyze these properties. We sequence genomes, whether eukaryotic or prokaryotic, with the latest next-generation sequencing technology and years of experience in this field. By tailor-made planning of the genome projects with subsequent data analysis and visualization through our bioinformatics team, you can obtain publishable sequence data on request. We are specialized primarily in de novo sequencing of Gram-positive bacteria (especially high GC-containing taxa) and fungi, but also re-sequencing is done professionally.
De Novo Sequencing
De novo sequencing refers to the primary decryption of the sequence of a previously unknown genome. Primarily methods are available to span long and many repetitive regions in the genome and to cover the genome as completely as possible. As a general rule, in a Whole Genome Shot project (WGS) read data from Nanopore (1D) and Illumina (Paired-End) were combined.
Example:
Complete and gapless genomes are the target in de novo sequencing projects. Long reads are a great advantage, because long reads span repetitive areas and simplify the assembly. Sequencing on the MiSeq in combination with long read data of the GridIon system are the most common approaches, This strategy has the advantage that a bacterial genome is directly gap-free after assembly and polishing in most of the cases. In additon, also eukaryotic genomes can be established until the chromosome level based on this strategy. Data from paired end or MatePair-only approaches, which allow the arranging of individual assembled contigs into larger scaffolds providing valuable information about the genome structure and are more cost-effective. Especially in organisms for which no reference genome is available described sequencing approaches are recommended.
Example of a genome project:
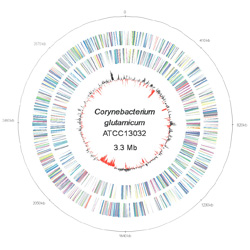
- Sequencing on the ONT GridIon and the Illumina MiSeq
+ Assembling the ONT
+ Polishing with Illumina data
+ Gap-free genome (very often otherwise refining with in silico methods)
+ Annotation
+ Comparative analyzes
+ Reconstruction of metabolic pathways
Resequencing
Genome re-sequencing is mainly used to detect genetic variations, such as SNPs, SNVs or indels, within eukaryotic populations or from different generations of prokaryotic production strains. By comparison with a reference genome, changes in the sequence and their consequences on phenotypes can be investigated.
Example:
For a resequencing project, a suitable reference genome is needed, to which the raw data are mapped. This approach requires a high genome coverage, which is achieved through a large number of individual reads. Depending on the size of the genome, the HiSeq 1500, MiSeq or NextSeq is suitable for this purpose.
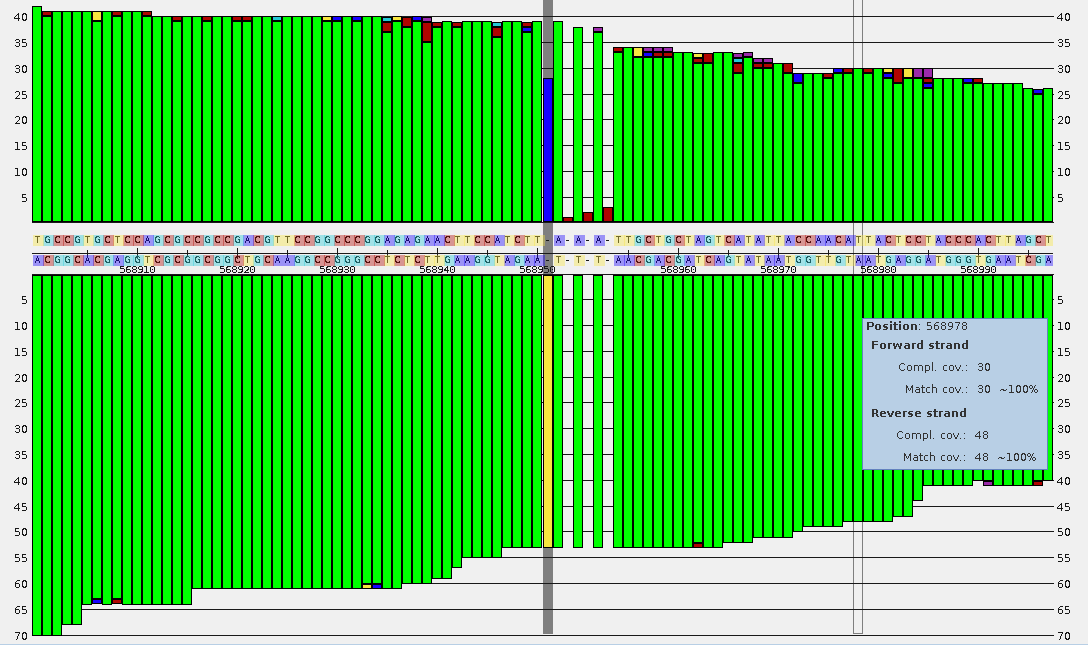 Example of a resequencing project:
Example of a resequencing project:
Sequencing on the HiSeq, NextSeq or on MiSeq
+ Read mapping
+ SNP and Indel analysis
To evaluate the genome sequences, various bioinformatic analysis methods are available. Take a look at our bioinformatics offer.