Metagenomics analysis can be done read-based or assembly-based. Read-based analysis of metagenomic datasets is a very computationally intensive application. The powerful infrastructure of the IIT GmbH allows automatic evaluation of even large metagenomic datasets. For the analysis of your metagenomic datasets, we use a set of established programs and databases used for the creation of taxonomic profiles (e.g., LCA, MetaCV, RDP, etc.) or for functional classification (e.g., COG, Pfam). We also offer static analysis for their metagenomic data sets. Your results are available through a client-server system.
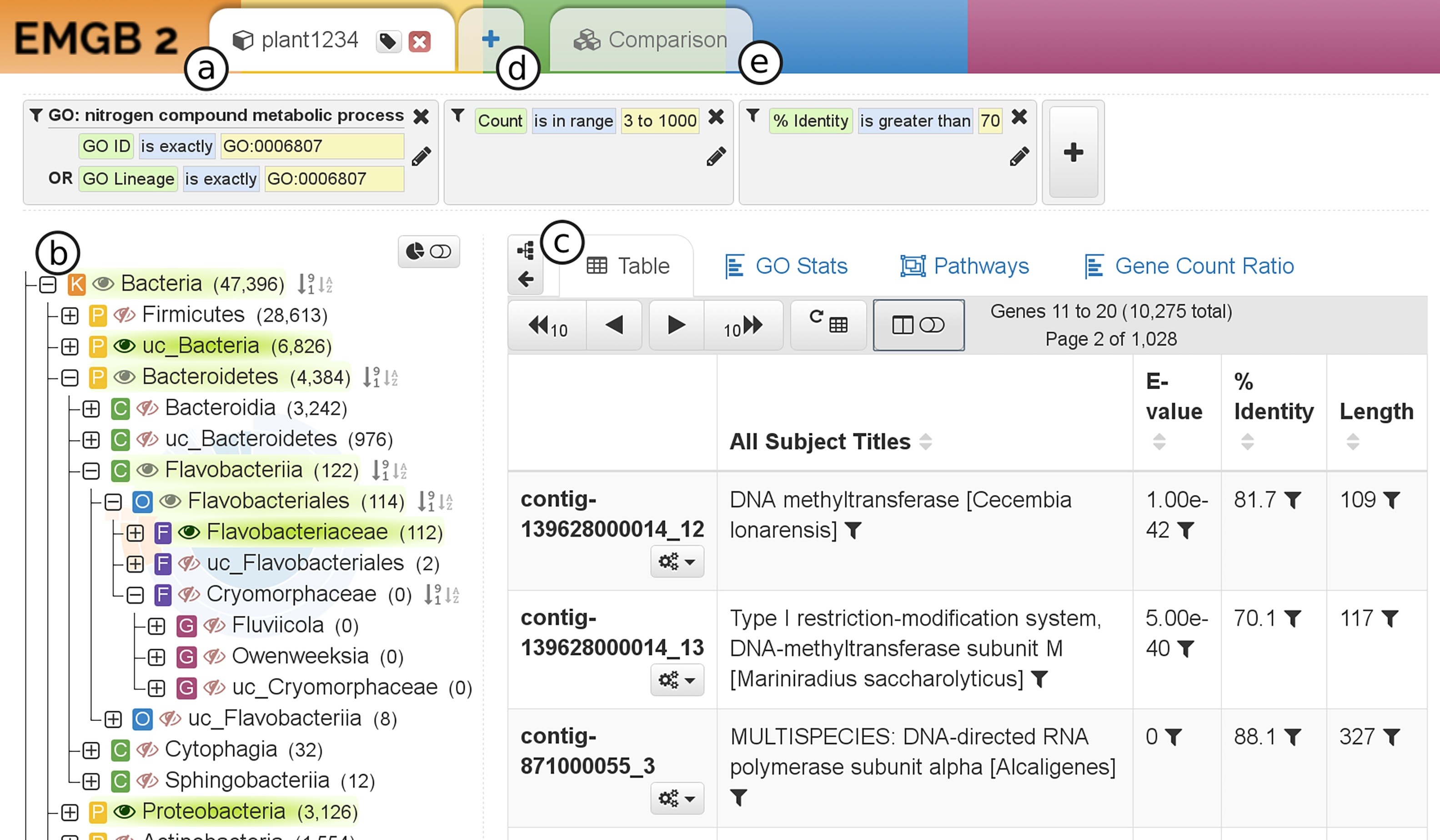
Assembly-based workflows attempt to assemble the reads from one or more samples, group (bin) the contigs from these samples into draft metagenome-assembled genomes (MAGs), then analyze the genes and contigs. Here, we provides pipelines to assemble, to group and to analyze metagenome data and assemblies. The IIT GmbH offers a responsive web interface to visually inspect the data. Our web application is especially built for browsing large annotated metagenomic datasets in real time and features an interactive phylogenetic tree, live GO term statistics as well as dynamically highlighted KEGG pathways for each dataset. In addition, a dedicated comparative viewer displays aggregated versions of these visualizations to draw comparisons across many different datasets at once.
Our range of metagenomic analyzes is complemented by our automated amplicon analysis, which offers you the analysis of taxonomic profiles and statistical calculations for their amplicon datasets.